
Journal of Artificial Intelligence in Bioinformatics
ISSN: request pending (Online) | ISSN: request pending (Print)
Email: [email protected]

In the field of contemporary life sciences, the intelligent design and optimization of nucleic acid sequences have become a hot issue in research, among which the design and optimization of nucleic acid sequence design are particularly critical and at the core, which is not only one of the core issues in the fields of synthetic biology, gene editing, and bio-informatics but also a key link in the development of life science and technology. The fundamental goal is to design and generate sequences that meet the multiple requirements of function, stability, and synthesizability according to the specific application needs, to promote the progress of genome engineering, drug development, and precision medicine [1]. In recent years, nucleic acid sequence design technology has made significant progress, especially in tool development, algorithm optimization, and application expansion [2, 3, 4]. Through the introduction of artificial intelligence algorithms and automated design platforms, nucleic acid sequence design has achieved efficient and intelligent closed-loop optimization, significantly improving the efficiency of sequence-function prediction and optimization [5].
Nucleic acid sequence design is widely used in synthetic biology [6], precision medicine [7], agriculture and food [8], industrial production [9] and information storage [10], etc. In synthetic biology, nucleic acid sequence design not only optimizes the efficiency of gene synthesis, but also accelerates the development of synthetic gene circuits and biological components, and promotes the development of fields such as biomanufacturing and environmental protection [11, 12, 13]. Meanwhile, the introduction of deep learning and artificial intelligence has brought revolutionary breakthroughs in nucleic acid sequence design, improving the accuracy and efficiency of gene expression efficiency, stability prediction, and sequence optimization [14, 15, 16]. With the development of high-throughput screening technology, experimental data-driven models have led to significant improvements in the accuracy and speed of functional verification of nucleic acid sequence design [17]. The rapid development in the field of information storage has also opened up new application prospects for nucleic acid sequence design [18].
With continuous technological innovations, nucleic acid sequence design will play an increasingly important role in the future and drive significant changes in related fields. This review explores intelligent design and optimization methods for nucleic acid sequences, focusing on existing technologies and methods, as well as future challenges and development directions. The paper first reviews the basic theories of nucleic acid sequence design, discussing the structural features, functions, and importance of DNA sequences in gene expression, which lays the foundation for the subsequent chapters. It then introduces traditional rule-driven DNA sequence optimization methods, focusing on strategies based on biological laws and chemical constraints, while also addressing their limitations in practical applications. Next, the paper explores intelligence-driven optimization methods, including generative models such as Variable Auto-Encoders, Generative Adversarial Networks, and Diffusion models, as well as optimization techniques based on large language models. These AI-driven approaches offer greater flexibility and accuracy for handling complex design tasks, overcoming some of the limitations inherent in traditional methods. The paper then discusses the practical significance of nucleic acid sequence design, along with the challenges involved in structure prediction. Finally, it summarizes the current challenges in nucleic acid sequence design and presents potential directions for future development, highlighting how intelligent algorithm-based approaches can drive further innovations in biology and medicine.
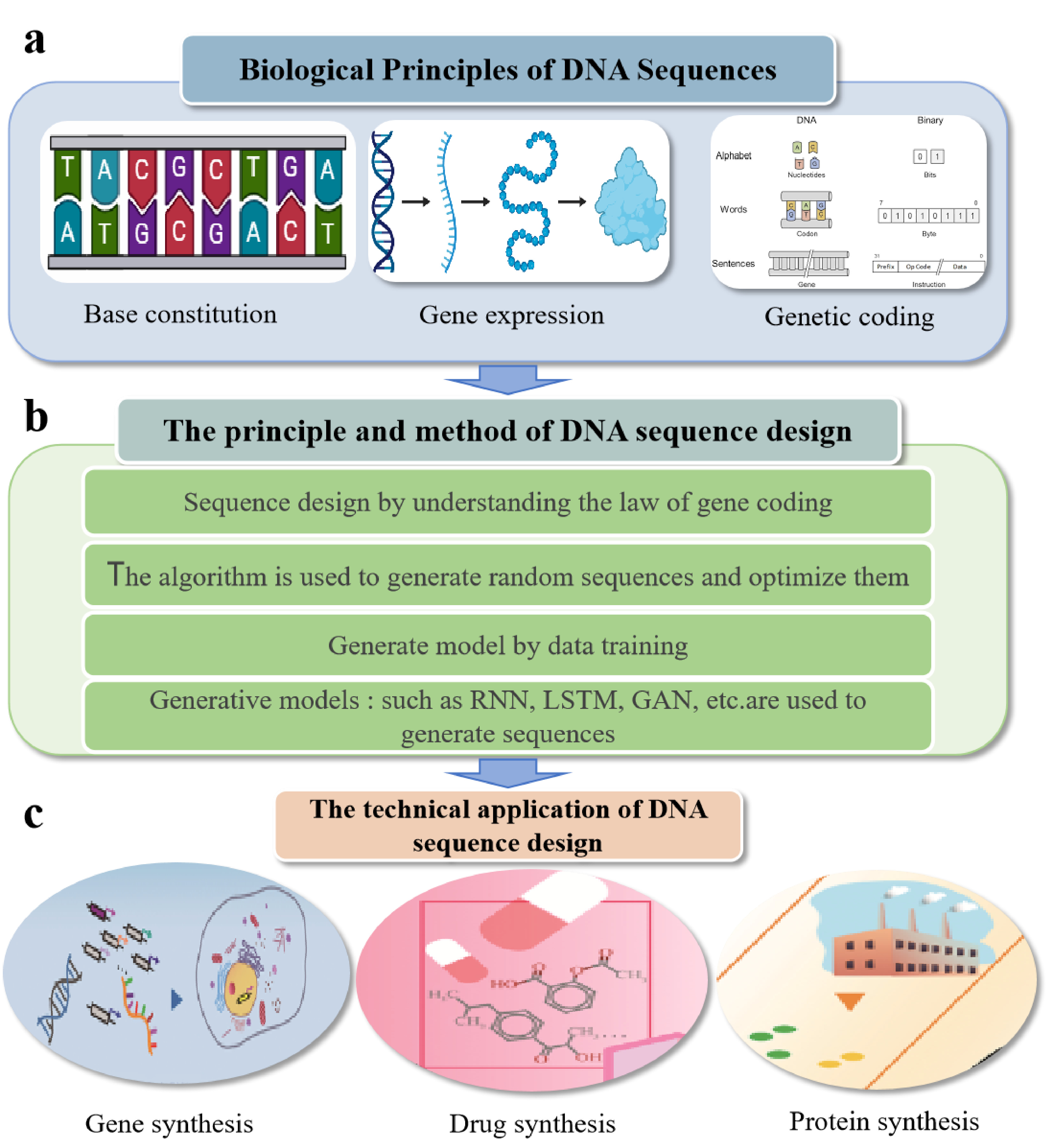
DNA sequence design and optimization aims to accurately construct DNA sequences according to specific functional requirements. This process involves the basic theories of molecular biology, computational biology, optimization algorithms, and artificial intelligence. Therefore, understanding these basic theories is crucial for effective DNA sequence design and optimization [19, 20]. DNA, as a carrier of genetic information, consists of four nucleotides—adenine (A), thymine (T), cytosine (C), and guanine (G)—which are arranged in a specific order to form a double helix structure. The nucleotide order determines the transmission of genetic information and the expression of genes [21, 22]. In addition to encoding proteins, DNA also plays a crucial role in various biological functions, such as regulating gene expression and protein synthesis [23, 24]. Therefore, when designing DNA sequences, several basic principles need to be followed. Firstly, to ensure the realization of gene function, the designed sequence should meet specific biological needs, such as encoding specific proteins or realizing gene regulatory functions [25, 26]. Secondly, high expression efficiency must be ensured, so that the expression level of the target genes meets experimental requirements [27]. The expression level of DNA is influenced by various factors. These factors include promoter strength, codon usage bias, RNA secondary structures, mRNA stability, and compatibility with the host cell's transcriptional machinery. Therefore, the designed sequence must not only meet functional requirements but also optimize these factors as much as possible to improve the efficiency and stability of gene expression [28]. In addition, the feasibility of the synthesis process (e.g., avoiding repetitive sequences or high GC regions) and compatibility with the host cell's genetic background (e.g., codon usage bias, presence of restriction enzyme sites, and promoter or terminator compatibility) must also be considered during sequence design [29].
In the process of DNA sequence design and optimization, multiple optimization theories and methods are usually applied. The optimization goal is often not a single gene expression level but involves the balance and optimization of multiple goals, such as sequence stability, synthesis cost, transcription, translation efficiency, etc.[30]. At this time, multi-objective optimization methods are particularly important, which can find the best balance between multiple conflicting goals. For example, codon optimization can enhance both translation efficiency and mRNA stability, while adjusting sequence complexity (e.g., reducing homopolymeric runs) may lower synthesis difficulty without compromising functionality [31, 32]. Combinatorial optimization methods, such as genetic algorithms and Monte Carlo simulations, are often used to achieve multi-objective optimization. These methods iteratively evaluate sequence variants based on global parameters (e.g., GC content, thermodynamic stability) and dynamically balance conflicting objectives [33]. Meanwhile, DNA sequence design is often accompanied by constraints such as sequence length, secondary structure, and gene regulatory element limitations [34], which increase the complexity of the design problem, and thus constrained optimization methods have emerged, which help to find the optimal or near-optimal solution under the premise of satisfying biological or engineering constraints. Overall, DNA sequence design is a complex, multi-faceted problem requiring integrated optimization approaches under biological constraints. As illustrated in Figure 1 [35], the theoretical foundation, design process, and application of DNA sequences are presented.
Traditional rule-driven DNA sequence optimization methods rely on biological principles and empirical laws to optimize gene expression, stability, and synthesizability by adjusting key elements (e.g. codons, GC content, secondary structure, etc.) in the gene sequences [36, 37, 38]. The core idea of these methods is to avoid some negative factors affecting gene expression or synthesis efficiency through the fine design of DNA sequences without relying on complex machine learning or artificial intelligence algorithms.
Codon optimization is a common strategy to enhance gene expression levels [39]. Codon preferences for the same amino acid vary across organisms, so by replacing codons in the target gene that do not match the host cell's tRNA usage preferences, translation efficiency can be significantly improved, thus enhancing the gene expression level [38, 40]. While codon optimization improves expression, excessive replacement of rare codons (over-optimization) may disrupt, over-optimization (e.g., eliminating all rare codons) may inadvertently disrupt co-translational protein folding or mRNA stability [41]. Secondly, rational optimization of GC content is also an important aspect of DNA sequence design. Excessively high or low GC content can lead to difficulties in DNA synthesis or sequence instability, so it is a commonly adopted optimization method by adjusting the GC ratio to avoid regions of extreme GC content [38, 42]. Recent studies suggest combining GC optimization with thermodynamic stability calculations (e.g., using mfold) to balance synthesis feasibility and mRNA function [43]. In addition, repetitive sequences and low-complexity regions in DNA sequences (e.g., long homopolymers of T or A and regions rich in single bases) can affect the effectiveness of PCR amplification and may lead to instability of the expression system [44, 45, 46]. For example, the elimination of poly-A/T tracts longer than six nucleotides can reduce polymerase slippage errors during the synthesis process [47]. Therefore, the design of these regions needs to be avoided during optimization to ensure gene stability. The design of promoters and regulatory elements is also crucial [48]. By rationally selecting the strength of promoters and optimizing the location of ribosome binding sites (RBS), transcription and translation efficiency can be effectively enhanced, thereby increasing the level of gene expression [49, 50]. However, promoter optimization must align with host RNA polymerase specificity (e.g., T7 promoters in E. coli) to avoid transcriptional incompatibility [51]. In addition, secondary structures of DNA sequences (e.g., hairpin structures, pseudoknots, stem-and-loop structures, etc.) can affect the translation process of genes and even lead to the stagnation of gene expression [52, 53]. Therefore, avoiding or reducing the formation of these unfavorable secondary structures is another key strategy to optimize gene expression [54]. For instance, stable secondary structures (G <-5 kcal/mol) are minimized using tools like NUPACK, while weak hairpins in 5' UTRs may be retained to enhance mRNA stability [55, 56].
Traditional rule-driven DNA sequence optimization methods include a variety of strategies, each of which functions according to different optimization goals and application scenarios. The template variation method optimizes gene expression and stability by mutating existing template sequences, but it relies on the template itself, which may lead to the design of sequences that lack innovation in structure or function and cannot avoid the inherent bias of the templates [57, 58]; The random sampling method, on the other hand, generates and screens sequences on a large scale to find an efficient design, and although it is capable of exploring a wider range of possibilities, the search is less efficient due to combinatorial explosion (e.g., the number of possible variants for a 1 kb gene may exceed 1020) [59]. In contrast, AI-driven methods differ fundamentally in three key aspects: (1) Design Paradigm: AI models (e.g., Transformers) learn hidden rules from large-scale sequence-activity datasets, enabling multi-objective optimization (e.g., balancing translation efficiency and mRNA stability) [60]; (2) Innovation Potential: AI generates non-natural sequences (e.g., synthetic promoters with less than 40% sequence similarity to natural sequences) that bypass template limitations [61]; (3) Efficiency: Compared to traditional random sampling, AI-driven virtual screening can reduce the number of candidates for experimental validation by over 90% [62]. For example, the Linear Design algorithm optimizes mRNA secondary structures 10,000 times faster than manual methods while maintaining codon adaptation [63]. These advancements highlight the complementary roles of traditional and AI-driven approaches in advancing synthetic biology.
Generative models can generate new sequences similar to real sequences by learning the statistical properties of DNA sequences [64, 65]. In addition to data generation, generative models can also enable dimensionality reduction by mapping the data space to the latent space, as well as predictive tasks by utilizing this learned mapping or supervised/semi-supervised generative model design [66, 67].
In the field of machine learning-driven DNA sequence design, generative models have gradually received widespread attention due to their potential to explore complex sequence spaces and discover novel design solutions. Variable Auto-Encoder (VAE) [68], a classical probabilistic generative model consisting of an encoder E and a decoder D, demonstrates unique advantages in sequence design tasks by its effective latent space characterization and flexible data generation capabilities. VAE combines the nonlinear representation capabilities of deep learning with the generative properties of probabilistic models, providing a powerful tool for biological sequence optimization and functional prediction [68, 69]. In DNA sequence design workflows, the VAE converts the input DNA sequences (usually represented by one-hot encoding or tokenized) into the mean and variance of a probability distribution (e.g., Gaussian distribution) in the latent space using an encoder [70]. The latent vector is then obtained by sampling from this distribution and passed to the decoder, which maps the latent vector back into the DNA sequences space to generate a new DNA sequence [71]. The training goal of the VAE is to minimize the reconstruction error and the KL divergence between the latent space distribution and a prior distribution, thereby learning a structured latent space capable of generating sequences similar to the original data [68]. Through this approach, VAE can generate DNA sequences that meet specific functional requirements, such as gene optimization and expression regulation. Figure 2(a) illustrates this application process.
The VAE demonstrates significant potential in biological sequence design, particularly excelling in functional sequence generation and stability optimization [71]. Its core strength lies in enabling controlled generation through probabilistic modeling of the latent space—researchers can efficiently learn latent representations of DNA sequences and sample the latent space to generate optimized sequences based on specific objectives (e.g., high stability) [69]. For instance, Sadeghi et al. [72] utilized VAE to design DNA-stabilized silver nanoclusters, enhancing sequence stability and functionality through automated feature extraction. Hawkins-Hooker et al. [71] generated functional protein variants, validating VAE's efficiency in protein design. Meanwhile, Greener et al. [73] and Moomtaheen et al. [74] applied VAE to metalloprotein design, novel protein fold exploration, and nanomaterial optimization, highlighting its versatility in biomolecular functional innovation. These studies demonstrate that VAE not only generates function-specific sequences but also provides systematic support for sequence stability, synthesizability, and cross-scale optimization.
Generative Adversarial Network (GAN) is a generative model composed of a generator (G) and a discriminator (D), first proposed by Goodfellow et al. [75] in their seminal work. In DNA sequences design, the generator (G) takes a noise vector z as input and generates a new sample G(z) as output [76]. In other words, the generator is responsible for mapping the potential space to the data space. The discriminator (D), on the other hand, takes as input a sample x and outputs a probability value D(x), which is used to evaluate whether x originates from a real data distribution or is synthesized by the generator G. The two networks are trained by adversarial training. These two networks optimize each other using adversarial training, with the discriminator D aiming to maximize the probability of correct classification, while the generator G attempts to confuse the discriminator by minimizing its probability of misclassifying the generated sample G(z) Figure 2(b) [66]. Essentially, this adversarial mechanism forms a zero-sum game until an equilibrium is reached, where the discriminator D is unable to tell whether G(z) originates from the true distribution or not.
The application of GAN in DNA sequence design has made significant progress in recent years, especially in gene expression optimization, synthetic biology, and new molecule design [77]. By using GAN, researchers can generate DNA sequences that meet specific needs, regulate gene expression, optimize synthetic processes, and even create artificial genomes [78]. Moreover, GANs have an edge in generating sequences with high fidelity, functionality, and preservation of complex structures, especially in tasks that require integration with experimental validation, such as protein design or data augmentation [79, 80]. Yu et al. [81] proposed the MichiGAN model, which samples single-cell data using generative adversarial networks and demonstrates the application of GAN in biological data generation. Yelmen et al. [82] used generative neural networks to successfully create an artificial human genome, further driving innovation in DNA sequence design. Zrimec et al. [14] regulated DNA sequences through deep generative design to optimize gene expression and demonstrated the application of GAN in gene expression regulation. In addition, MedGAN proposed by Macedo et al. [83] optimized GAN in combination with Graph Convolutional Networks(GCN) for generating new molecular structures, further expanding the application of GAN in DNA sequence design and molecule generation. Through these studies, GAN provides a revolutionary technological pathway for DNA sequence design and gene expression optimization, which promotes the development of fields such as precision medicine, gene editing, and synthetic biology.
Diffusion models (DM) are a class of generative models that have achieved remarkable success in recent years in generative tasks, especially in the fields of image generation, speech generation, and sequence generation. The basic idea is derived from the diffusion process in non-equilibrium thermodynamics [84], where the model simulates a forward stochastic differential equation (SDE) to progressively add noise to data, followed by a reverse SDE to recover the original data through iterative denoising. Unlike VAEs and GANs, DM generates DNA sequences by explicitly modeling the sequential corruption and reconstruction of sequence distributions [85]. Specifically, the diffusion model first performs a 'noise addition' process on the input DNA sequence, which gradually transforms it into a random noise sequence [86], and then gradually denoises it through a learned back-diffusion process to recover a DNA sequence that meets the functional and structural characteristics of the target Figure 2(c) [87]. The strengths of diffusion models stem from their ability to model high-dimensional correlations through the progressive denoising mechanism, as well as their flexible conditional control interfaces, which endow them with broad application prospects in the fields of precision medicine and synthetic biology [85, 88].
In recent years, DM has been gradually applied to DNA sequence design as an emerging method for generating and optimizing gene sequences. The potential diffusion model proposed by DaSilva et al. [85] provides an innovative framework for DNA sequence generation, which generates DNA sequences by mapping them into the potential space and accurately controls the properties of the generated sequences through an optimization process. Sarkar et al. [86] used the discrete diffusion model to design DNA sequences with modifiable activities, providing a new design tool for gene regulation and synthetic biology. Wang et al. [89] proposed the AptaDiff model specifically for the design of aptamers and used the diffusion model for the de novo design and optimization of novel aptamers, which effectively improved the targeting and specificity of the sequences. In addition, the Dirichlet diffusion model introduced by Avdeyev et al. [90] provides a new theoretical basis for biological sequence generation, which is based on the Dirichlet process to optimize the sequence generation process, thus enhancing the diversity and biological functions of the generated sequences. Through these studies, the diffusion model provides a powerful generative capability for the design and optimization of DNA sequences, especially showing great potential in the fields of gene expression regulation, aptamer design, and bioinformatics.
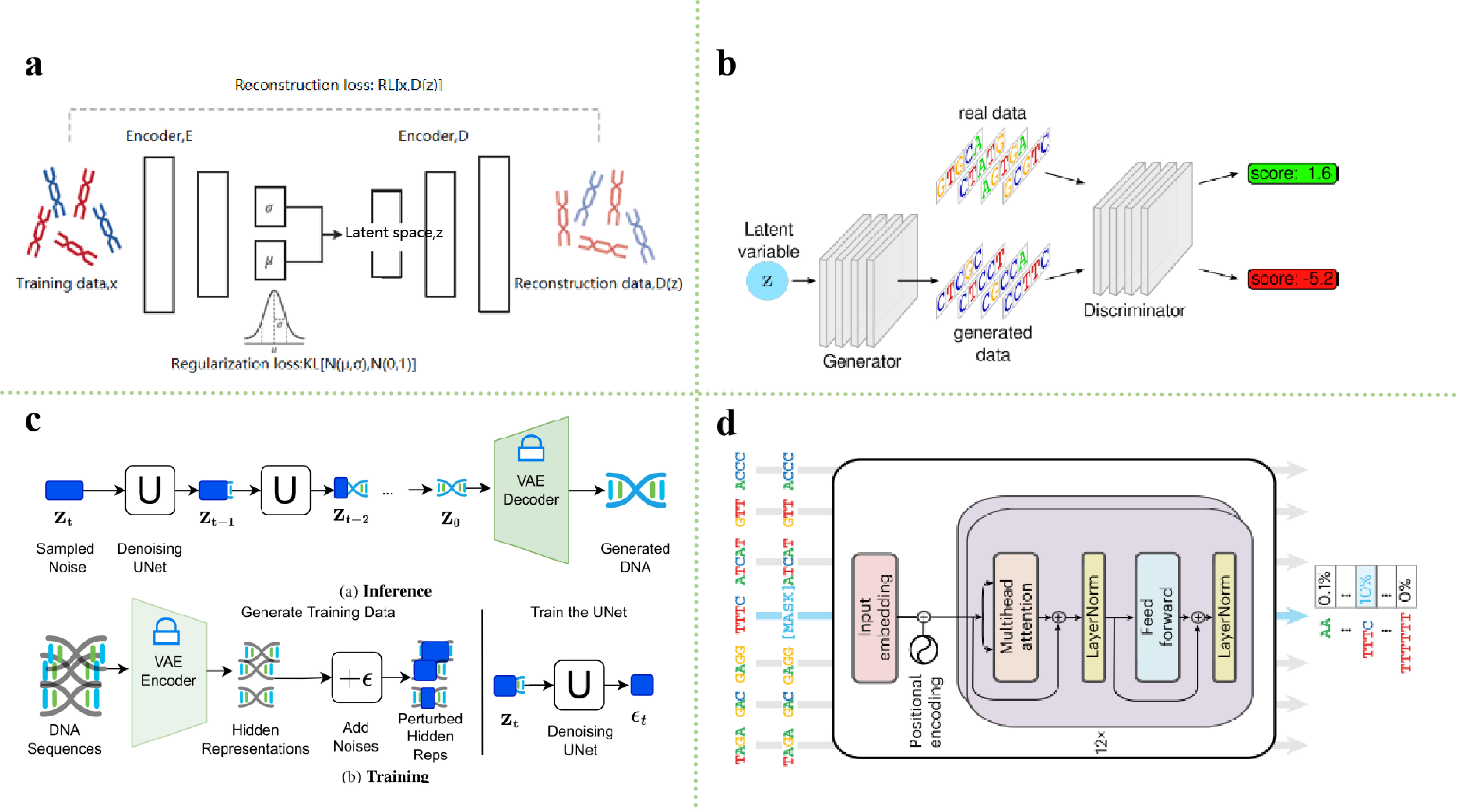
With the emergence of large-scale pre-trained language models, especially in the field of Natural Language Processing (NLP), many researchers have started to draw on the principles of language models to deal with genomic data and develop genomic language models [91]. The basic goal of language models is to predict the next word in a given context by learning the relationship between individual words in a language [92, 93]. In NLP, language models are usually trained based on large amounts of textual data to learn how to predict the next word based on the previous word, thus generating coherent sentences [94]. Analogous to DNA sequence design, DNA sequences can also be viewed as a form of "language," where 'words' can be base pairs (A, T, C, G) or vocabularies obtained through other tokenization techniques (such as K-mer, One-hot encoding) [95, 96]. The model learns the relative positions and order of bases or tokens in various ways, enabling it to generate reasonable and biologically meaningful DNA sequences, as shown in Figure 2(d) [97]. The core advantage of language models stems from the ability of the self-attention mechanism to model long-range dependencies and the multimodal conditional control interface, which has opened up a new paradigm for precision medicine and synthetic biology [98, 99].
Language models have been widely used in the design and analysis of DNA sequences, especially in the fields of genomics and synthetic biology. By using linguistic models, researchers can deeply understand the 'language' of DNA sequences and generate DNA sequences that meet specific needs. For example, Ji et al. [100] proposed the DNABERT model, based on the bidirectional encoder representation (BERT) architecture, which was successfully applied to the pre-training of DNA sequences and demonstrated its powerful language model ability in genome sequence analysis. Shao et al. [101] developed a long context language model for decoding and generating the bacteriophage genome, which provides a new idea for genome sequence generation. Nguyen et al. [13] proposed the Evo model, which combines molecular to genomic-scale sequence modeling and design, demonstrating its potential for large-scale genomic analysis and providing new insights for gene design. Recently, Madani et al. [102] successfully generated functional protein sequences of multiple families using a large-scale language model. These studies show that language models provide powerful theoretical and technical support for the design of DNA sequences, and can help predict gene function, optimize gene expression, and promote the further development of genomics.
Nucleic acid sequence design plays a crucial role in nucleic acid structure prediction. Through in-depth analysis of nucleic acid sequences, researchers can identify potential structural features, folding rules, and functional regions, providing powerful support for structure prediction. In particular, methods such as evolutionary information, multiple sequence comparison, covariance analysis, and secondary structure prediction can significantly improve the accuracy of structure prediction [103]. Meanwhile, sequence analysis models based on machine learning and deep learning can automatically learn the complex relationship between sequence and structure from a large amount of data, further promoting the development of nucleic acid structure prediction technology. For instance, Aslam et al. [104] demonstrated how adaptive machine learning frameworks enhance predictive accuracy in biological systems through domain-specific optimization. With the continuous enrichment of datasets and technological advances, nucleic acid sequence analysis will play a more important role in nucleic acid structure prediction in the future.
The functions of nucleic acids (e.g., catalysis, regulation, binding ligands, etc.) often depend on their 3D structures; therefore, predicting the 3D structures of nucleic acids can help design sequences with specific functions, enhance the accuracy of the design, and advance the understanding of the functions of nucleic acids, as well as optimize the process of sequence analysis, improve the accuracy of comparisons, support the discovery of drug targets, and guide experimental design and genetic engineering. Butt et al. [105] highlighted the integration of intelligent classification models in biomedical applications, which aligns with the need for precision in functional nucleic acid design. Structure prediction promotes a comprehensive understanding of nucleic acid sequences by providing insightful structural context for sequence analysis. DNA or RNA sequences in the genome form specific three-dimensional structures when folded, and these structures determine molecular interactions and biological functions. By accurately predicting these structures, it can help to design more functional molecules, rather than just predicting their basic function based on sequence. For example, in nanobiotechnology, self-assembling DNA or RNA molecules are designed to form specific nanostructures or nanomachines [106]. Rasool et al. [107] further exemplified this by developing a DNA-based file storage system optimized for medical data, demonstrating the synergy between sequence design and structural stability. Using 3D structure prediction, sequences with predetermined structures can be designed and their stability and functionality in practical applications can be ensured. Additionally, Rasool et al. [108] proposed a strategy-based optimization algorithm for DNA data storage encoding, underscoring the importance of sequence-structure co-design in emerging technologies. The two complement each other along with various deep learning approaches paving the way for each other.
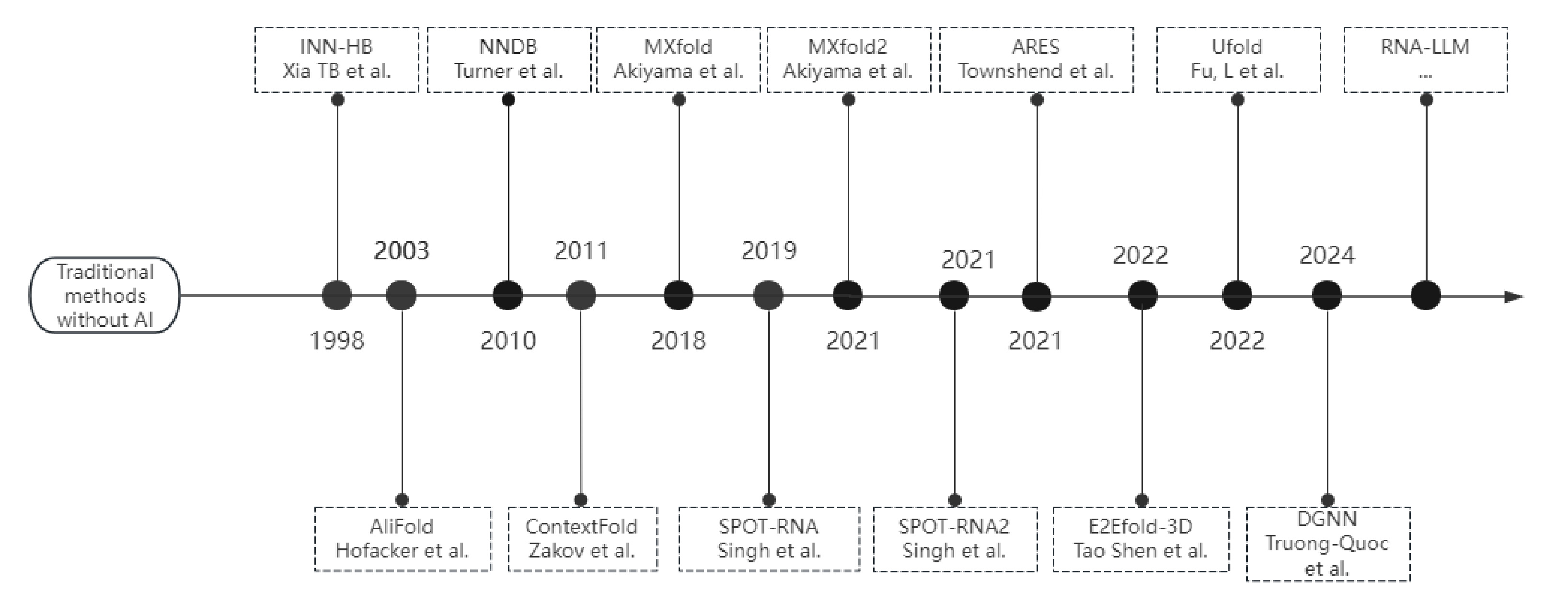
At the end of the 20th century, Westhof et al. [109] used molecular mechanics and molecular dynamics simulation software (e.g., AMBER, GROMOS, Xplor, etc.), which are based on classical physical models and algorithms to simulate the changes in nucleic acid structure by calculating the interaction energies between atoms. Multiple sequence comparison tools, such as CLUSTAL, were used to find similarities between sequences based on dynamic programming algorithms or heuristic algorithms. Subsequently, in the 21st century, methods based on comparative sequence analysis such as the RNAfold method [110] are limited by arithmetic power and algorithms and are not accurate enough in predicting complex RNA secondary structures. It is extremely difficult to fully understand the RNA folding mechanism. While data-driven methods are powerful in scenarios with limited mechanistic understanding, models integrating domain knowledge (e.g., thermodynamic rules or evolutionary conservation) often achieve superior interpretability and performance. For instance, hybrid approaches combining deep learning with biophysical principles have advanced RNA structure prediction [111]. These methods can learn the underlying folding patterns from large amounts of training data. Over the past decades, machine learning and deep learning methods have been used in many aspects of RNA secondary structure prediction methods to improve prediction performance. Figure 3 illustrates the development of nucleic acid structure prediction in the AI era.
RNA secondary structure prediction methods based on machine learning and deep learning typically learn functions that map inputs (features) to outputs by tuning model parameters based on known input and output pairs. Many of them employ free energy parameters, encoded RNA sequences, sequence patterns, or evolutionary information as key features, and their outputs can be categorical labels (e.g., paired or unpaired) or continuous values (e.g., free energy). When new inputs are provided to a trained model, the model can classify the corresponding labels or predict the corresponding values [112]. The nearest neighbor model (NNDB) developed by Turner [113] was an early and fairly commonly used approach, which provided a fairly accurate approximation of RNA free energy. However, several thermodynamic parameters of the NNDB model had to be based on a large number of optimal melting experiments, which were both time-consuming and labor-intensive [55, 114], and due to the associated technical difficulties, not all free energy changes in the structural elements could be measured. Due to the difficulty of obtaining the relevant parameters, several machine-learning techniques have been used to optimize the parameters in energy models. These techniques can use fine-grained models that estimate fractions using known thermodynamic data or RNA secondary structure data to obtain richer and more accurate representations of the features. Xia et al. [115] first trained a linear regression model using known thermodynamic data to infer some of the thermodynamic parameters and extended the neural network model to a more accurate model, the INN-HB model. This model provides a better fit for known experimental data. However, a disadvantage of this approach is that the parameters of some structural elements are fixed before other parameters are calculated, which limits the range of possibilities for the entire parameter set.
Although machine learning-based free energy parameter methods have successfully improved the accuracy of RNA secondary structure prediction, the energy model is still far from ideal. The machine learning-based parameter estimation methods can only replace some wet lab experiments aimed at obtaining energy parameters. As a result, Zakov et al. [116] proposed the ContextFold tool, which not only relies on traditional energy parameters but also takes into account the contextual information in the RNA structure, significantly improving the accuracy and flexibility of RNA structure prediction. Later, Akiyama et al. [117] integrated thermodynamic methods with SSVM and developed MXfold, which overcame the limitations of traditional tools in energy model optimization and large-scale data processing, but had limited prediction accuracy for long-stranded RNAs (e.g., mRNAs or lncRNAs) and did not support the prediction of pseudo-knots. Sato et al. [118] developed MXfold2 to overcome the above-mentioned limitations, marking a new level of performance in RNA structure prediction.
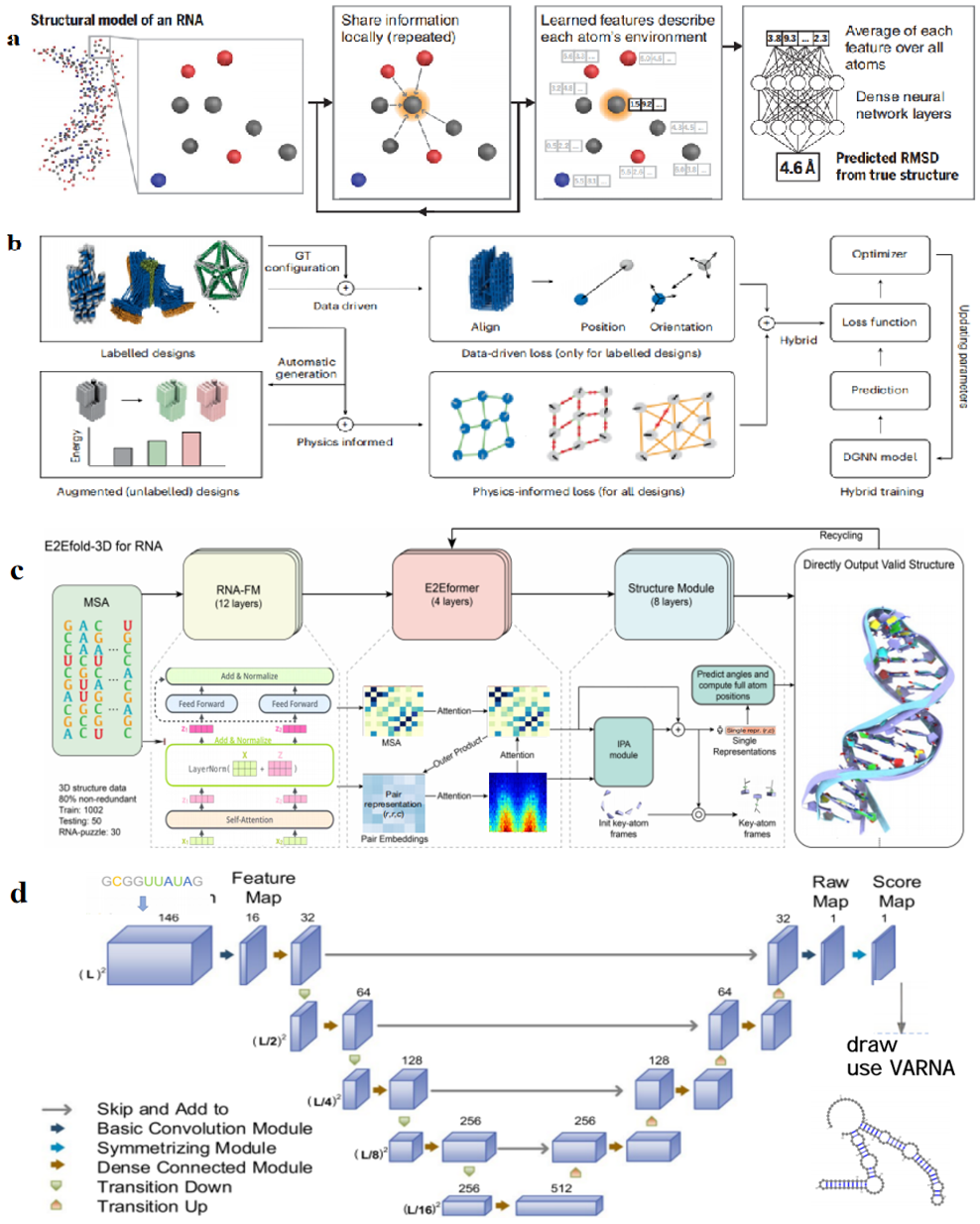
Since 2020, more and more models for predicting the 3D structure of nucleic acids based on deep learning methods have appeared in the public eye with innovations in technologies related to deep learning and nucleic acid sequence design. Linyu Wang's team proposed a new method called DMfold [119] based on Bi-LSTM to predict the secondary structure of RNA containing pseudoknots, which combines deep learning and the improved base pair maximization principle, fully absorbing the advantages and avoiding some of the disadvantages of the multi-sequence method (MPM) and the single-sequence method (SPM), but DMfold does not achieve the optimal accuracy in the case of known However, the accuracy of DMfold is not optimal when the sequence is known to be insufficient, and with the continuous optimization of sequence design technology, DMfold will gain more space for development. Kunitski et al. [120] proposed the first end-to-end deep learning model SPOT-RNA to predict the secondary structure of RNA.SPOT-RNA treats RNA secondary structure as a CT table and uses an ultradeep hybrid network that combines ResNet and 2D-BBN to predict the secondary structure of RNA. SPOT-RNA treats RNA secondary structures as CT tables and uses an ultra-deep hybrid network that combines the ResNet and 2D-BLSTM networks to make predictions, with the ResNet network capturing contextual information across the entire sequence and the 2D-BLSTM efficiently propagating long-range dependencies in RNA structures. Through migration learning, SPOT-RNA can achieve good results even with limited samples. Experimental results showed that SPOT-RNA performed well on multiple RNA benchmark datasets, and in a follow-up development, the SPOT-RNA2 model was proposed by the same research group [121]. This model employs evolutionarily derived sequence profiles and mutation coupling as inputs, uses the same migration learning approach, and outperforms SPOT-RNA in all types of base pair prediction. Shen et al. [122] developed the E2Efold-3D model, as shown in Figure 4(c), which is another deep learning for predicting RNA secondary structure, E2Efold-3D is another method that can directly predict the 3D structure of RNA without external templates, which greatly improves the accuracy of RNA structure prediction through secondary structure-assisted self-distillation, multidimensional information fusion, and joint training, especially in addressing data scarcity and structural complexity, showing its unique advantages. It is worth mentioning that E2Efold uses a deep learning approach to achieve end-to-end RNA structure prediction by obtaining binary classification scoring matrices of base pairs from input RNA sequences, whereas the ARES model, as shown in Figure 4(a), which is different from the classification model E2Efold, is a regression model based on geometric deep learning developed by Townshend et al. [123] only trained a new RNA tertiary structure scoring model from 18 known RNA tertiary structures published between 1994 and 2006. The input to ARES is the 3D coordinates and chemical element type of each atom, and the output is the root-mean-square deviation (RMSD) between the predicted structural model and the true structure, which means that ARES learning is completely featureless without any predefined features, and the model directly learns and extracts features from the raw data, effectively reducing human bias and significantly excelling in the discovery of new features. ARES significantly outperforms other scoring functions and models despite using a limited number of known RNA structures. The REDfold model, as shown in Figure 4(d) [124], which uses a CNN-based encoder-decoder network to learn the dependencies in RNA sequences and effectively propagates information across layers through symmetric skip connections, achieves better performance in both efficiency and accuracy for RNA secondary structure prediction. Another RNA prediction model, Ufold, instead of directly inputting the 3D coordinates and chemical element type of each atom, inputs a matrix of all possible base pairs (canonical and non-canonical base pairs) and pairing features of the RNA sequence. Ufold converts the input matrix and pairing features into base pairing probabilities for predicting the RNA secondary structure through the use of a fully convolutional network (FCN) [125].
The outstanding contribution of nucleic acid sequence design in structure prediction is not only in the direction of RNA prediction but also in the direction of DNA, Chien Truong-Quoc's team has developed a DNA-origami-based graph neural network (DGNN), as shown in Figure 4(b) [126], which is more focused on the instantaneous and accurate prediction of DNA origami structures. The authors' innovative hybrid data-driven and physically-guided training approach greatly alleviates the difficulty of training purely data-driven models for DNA origami design, as the dataset is very scarce, and the authors also integrate pre-trained models corresponding to various shapes of DNA origami, which enhances the adaptability and performance of the DGNN for various types of data. The DGNN also makes outstanding contributions in the areas of structure prediction of supramolecular assemblies and inverse design of DNA origami. DGNN has also made outstanding contributions in the areas of supramolecular assembly structure prediction and DNA origami inverse design. Recently, many large language models (RNA-LLM) for RNA structure prediction have appeared [127], and perhaps shortly, there may be new advances in the combination of nucleic acid structure prediction and large language models.
Despite significant progress made by intelligent design and optimization methods in the field of nucleic acid sequences, multiple technical bottlenecks and systemic challenges remain. Existing computational models can perform preliminary predictions using rule engines, machine learning, and generative approaches; however, considerable uncertainty persists when dealing with complex nonlinear systems such as gene regulatory networks [128, 129]. The experimental validation phase relies on high-throughput screening technologies, which are plagued by the high cost of equipment and consumables, thereby limiting the overall research and development efficiency of the "computation-experiment" dual-track validation model. Moreover, practical applications of artificial intelligence face three key constraints: computational resources, data quality, and ethical standards. Training deep generative models consumes enormous computational power—for instance, handling databases with millions of sequences requires terabyte-level storage and GPU clusters [130]; noise data in public nucleic acid sequence databases can account for as much as 30%, and the scarcity of data for certain disease-related targets severely undermines model reliability [131, 132]; additionally, the "black box" nature of AI may lead to issues such as untraceable decision-making and algorithmic bias. For example, in gene editing design, the absence of an interpretable mechanism might conceal potential off-target risks [133].
Nevertheless, AI technologies have demonstrated revolutionary empowerment potential across the entire nucleic acid sequence design chain. From RNA three-dimensional structure prediction algorithms expanded from AlphaFold3 to target-sequence matching systems incorporating Transformer architectures, AI has reduced traditional design cycles from months to weeks [134]. Furthermore, deep generative models can automatically generate candidate sequences that satisfy specific binding energy, stability, and functionality criteria; when combined with automated experimental platforms, they enable high-throughput synthesis and validation [16]. This closed-loop approach not only accelerates the development of antiviral nucleic acid drugs but also provides a new paradigm for the design of cancer vaccines and gene editing tools (such as CRISPR). However, the ultimate challenge of technological implementation lies in balancing the pace of innovation with risk management. Only by establishing a multidimensional governance framework that encompasses technological interpretability, data ethics, and public engagement can we ensure that AI truly advances nucleic acid sequence design toward precision and responsible innovation.
Future breakthroughs should focus on multimodal data integration and interdisciplinary collaborative innovation. By integrating multidimensional data from genomics, epigenetics, and proteomics, a more comprehensive framework for sequence function prediction can be constructed. At the same time, developing lightweight model architectures (such as those based on knowledge distillation) and distributed computing solutions is expected to lower the computational threshold, while the adoption of privacy-preserving techniques like federated learning can help alleviate data silo issues. On the ethical front, it is necessary to establish a transparent mechanism that spans the entire lifecycle of model design, training, and deployment—for example, by introducing linear artificial chromatography imaging methods to elucidate neural network decision paths and by supervising technological compliance through a dynamic ethics review committee.
The application of artificial intelligence in nucleic acid sequence design has already demonstrated enormous potential, yet its practical implementation still requires balancing the speed of innovation with risk management. Only by building a multidimensional governance system that includes technological interpretability, data ethics, and public participation can AI truly drive nucleic acid sequence design toward precision and responsible innovation. Through technological breakthroughs, expanded applications, and improved governance, AI is poised to usher in a new revolution in the life sciences, providing a powerful engine for human health and sustainable development.
 Copyright © 2025 by the Author(s). Published by Institute of Emerging and Computer Engineers. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
Copyright © 2025 by the Author(s). Published by Institute of Emerging and Computer Engineers. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. Journal of Artificial Intelligence in Bioinformatics
ISSN: request pending (Online) | ISSN: request pending (Print)
Email: [email protected]

Portico
All published articles are preserved here permanently:
https://www.portico.org/publishers/iece/